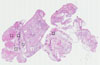
TACSTD2
Search result
|
TISSUE
PRIMARY DATA
URINARY BLADDER
ANTIBODIES
AND VALIDATION 
Dictionary
Urinary bladder
Tissue proteome
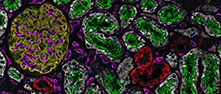
Tissue methods

|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 The Human Protein Atlas project is funded
The Human Protein Atlas project is funded