Search result
SUMMARY

TISSUE
BRAIN
SINGLE CELL
TISSUE CELL
PATHOLOGY
DISEASE
IMMUNE
BLOOD
SUBCELL
CELL LINE
STRUCTURE
INTERACTION
|
TISSUE
PRIMARY DATA
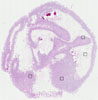
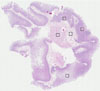
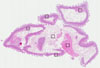
SMALL INTESTINE
ANTIBODIES
AND VALIDATION 
Dictionary
Small intestine
Tissue proteome
Intestine
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 The Human Protein Atlas project is funded
The Human Protein Atlas project is funded